PTM-DIA
DIA特异的翻译后修饰位点定位算法
相较于传统的DDA方法,DIA方法进行翻译后修饰的数据采集能够得到更加全面的信息比如碎片离子的完整同位素峰分布,生成与目标母离子峰型相似的色谱洗脱峰(XIC),而后者则有助于系统的去排除干扰碎片离子,这是传统DDA方法无法做到的。以上两个方面结合碎片离子强度及质量精度匹配进行鉴定评分,每个碎片离子将会得到特定的加权得分进一步用来计算特定位点的定位得分。
首先该算法会将识别到的肽段-子离子进行组合,针对每一特定的组合,将该修饰肽段所有可能存在的修饰位点组合一一列举(如图a),然后针对每一候选的修饰位点,该算法会将该组合内的所有子离子与母离子进行匹配,从而评判该碎片离子是确证了该修饰位点的存在(Confirming)还是能够否定(Refuting)该修饰位点的存在(图c),随即结合特征峰、质量准确性和XIC(图b)相关性等方面的匹配,计算每个碎片离子的得分。最后,该候选修饰位点的得分等于所有确证离子的得分总和减去否认离子的得分总和(Score(cand))。而该位点发生修饰的概率则为:所有包含该特定修饰位点的候选得分加和/全部修饰位点候选得分加和(如图d)
-

图a
-

图b
-

图c
-
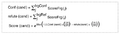
图d1
-

图d2




PTM位点水平定量输出
对于翻译后修饰蛋白质组的定量分析,用户最为关注的则是位点水平的变化,而现有的大多数搜库软件都只能报告肽段水平的定量值。众所周知,由于不可避免的漏切、非特异性酶切、可变修饰等因素,导致众多的磷酸化位点会同时以多种肽段形式被检测到(如下图f),以PX蛋白的T10和S15两个磷酸化位点为例,我们可能检测到针对这两个位点的如下5条磷酸化肽段,为了便于理解我们假设每条磷酸化肽段的定量值是100。
在SN15及以上版本中,软件除了会在原有的母离子及肽段水平报告相应定量值外,新增了PTM Site Report模块,以位点为单位展示和输出定量结果。
默认参数(如下图g)下位点定量结果的输出遵循以下原则:
- Localization Probability阈值设定为0.75,最终Report中报告的位点都是满足该阈值的修饰位点;
- 针对特定磷酸化位点,单磷酸化和多磷酸化状态的定量分开报告(Multiplicity,如果不区分单磷酸化和多磷酸化,则不勾选此参数即可。考虑到很多研究表明特定位点在不同的Multiplicity下生物学功能不同,建议采用默认参数;
- 针对特定磷酸化位点,其定量值为所有包含该修饰位点的母离子(见图h PTM.Group列)的定量值的总和(对应的参数:PTM Consolidation: Sum)
- Sequence window的划分,Flanking Region下可以指定划分窗口时以修饰位点为中心,向其前后各取几个氨基酸
-

图f SN进行位点整合流程示意图
-
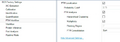
图g PTM Workflow默认参数设置
-
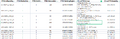
图h PTM Site Report示意图
参考文献
- Bekker-Jensen, D.B., Bernhardt, O.M., Hogrebe, A. et al. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat Commun 11, 787 (2020). https://doi.org/10.1038/s41467-020-14609-1